
Shopping Cart
- Remove All

Your shopping cart is currently empty

 Argentina
Argentina Australia
Australia Austria
Austria Belgium
Belgium Brazil
Brazil Bulgaria
Bulgaria Croatia
Croatia Cyprus
Cyprus Czech
Czech Denmark
Denmark Egypt
Egypt Estonia
Estonia Finland
Finland France
France Germany
Germany Greece
Greece Hong Kong
Hong Kong Hungary
Hungary Iceland
Iceland India
India Ireland
Ireland Israel
Israel Italy
Italy Japan
Japan Korea
Korea Latvia
Latvia Lebanon
Lebanon Malaysia
Malaysia Malta
Malta Morocco
Morocco Netherlands
Netherlands New Zealand
New Zealand Norway
Norway Poland
Poland Portugal
Portugal Romania
Romania Singapore
Singapore Slovakia
Slovakia Slovenia
Slovenia Spain
Spain Sweden
Sweden Switzerland
Switzerland Taiwan,China
Taiwan,China Thailand
Thailand Turkey
Turkey United Kingdom
United Kingdom United States
United States Other Countries
Other Countries| Pack Size | Price | Availability | Quantity |
|---|---|---|---|
| 50 μg | $320 | In Stock | |
| 100 μg | $567 | 7-10 days | |
| 200 μg | $1,000 | 7-10 days | |
| 500 μg | $2,120 | 7-10 days |
| Biological Information | Testing in progress |
| Description | GLA/alpha-Galactosidase A Protein, Human, Recombinant (His) is expressed in HEK293 mammalian cells with His tag. The predicted molecular weight is 46.8 kDa and the accession number is P06280. |
| Species | Human |
| Expression System | HEK293 Cells |
| Tag | C-His |
| Accession Number | P06280 |
| Synonyms | GLA/α-Galactosidase A Protein,galactosidase, α,galactosidase, alpha,GALA |
| Construction | The Human GLA (NP_000160.1) (Met 1-Leu 429) was fused with a polyhistidine tag at the C-terminus. |
| Protein Purity | > 97 % as determined by SDS-PAGE 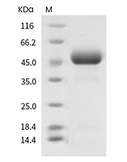 |
| Molecular Weight | 46.8 kDa (predicted) |
| Endotoxin | < 1.0 EU/μg of the protein as determined by the LAL method. |
| Formulation | Lyophilized from a solution filtered through a 0.22 μm filter, containing 50 mM Tris, 150 mM NaCl, pH 7.5. Typically, a mixture containing 5% to 8% trehalose, mannitol, and 0.01% Tween 80 is incorporated as a protective agent before lyophilization. |
| Reconstitution | A Certificate of Analysis (CoA) containing reconstitution instructions is included with the products. Please refer to the CoA for detailed information. |
| Stability & Storage | It is recommended to store recombinant proteins at -20°C to -80°C for future use. Lyophilized powders can be stably stored for over 12 months, while liquid products can be stored for 6-12 months at -80°C. For reconstituted protein solutions, the solution can be stored at -20°C to -80°C for at least 3 months. Please avoid multiple freeze-thaw cycles and store products in aliquots. |
| Shipping | In general, Lyophilized powders are shipping with blue ice. |
| Research Background | Alpha-galactosidase A, also known as Alpha-D-galactoside galactohydrolase, Alpha-D-galactosidase A, Melibiase and GLA, is a member of the glycosyl hydrolase 27 family. GLA is used as a long-term enzyme replacement therapy in patients with a confirmed diagnosis of Fabry disease. Defects in GLA are the cause of Fabry disease (FD) which is a rare X-linked sphingolipidosis disease where glycolipid accumulates in many tissues. The disease consists of an inborn error of glycosphingolipid catabolism. FD patients show systemic accumulation of globotriaoslyceramide (Gb3) and related glycosphingolipids in the plasma and cellular lysosomes throughout the body. Clinical recognition in males results from characteristic skin lesions (angiokeratomas) over the lower trunk. Patients may show ocular deposits, febrile episodes, and burning pain in the extremities. Death results from renal failure, cardiac or cerebral complications of hypertension or other vascular disease. Deficiency of GLA leads to the accumulation of glycosphingolipids in the vasculature leading to multiorgan pathology. In addition to well-described microvascular disease, deficiency of GLA is also characterized by premature macrovascular events such as stroke and possibly myocardial infarction. |

 Hello! How can I help you today?
Hello! How can I help you today? Copyright © 2015-2024 TargetMol Chemicals Inc. All Rights Reserved.